|
<< HOME
PlatinumCNV 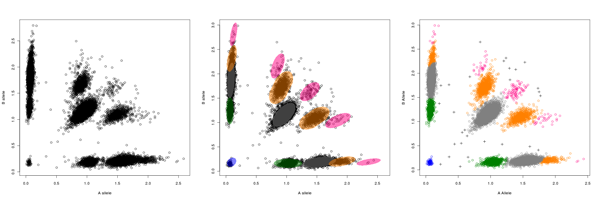 PlatinumCNV (Kumasaka et al., 2011) is a software package to fit a Bayesian Gaussian mixture model (GMM) on the SNP array signal intensity data, and infer individual's allele-specific copy number genotypes at genome-wide SNP loci in a SNP array. The software package currently consists of two executable programs, gmm and call; The gmm program fits the Bayesian GMM on the 2-dimensional (allele-specific) signal intensities, and the call program infers individual's genotypes. We also include additional R functions to plot the results of gmm and call. Latest Version: ZIP | TGZ MAKE AND INSALL Download one of the above archive files (both are the same), and extract the package directory. PlatinumCNV is written in C language and using a part of blas and lapack routines. Before you make the package, you need to make sure that those routines are available in your computer. If lapack and blas libraries are installed in /usr/local, the following commands make the package: % tar zxvf PlatinumCNV_* % cd PlatinumCNV_*/src % make % make test If the package was compiled successfully, you may find gmm and call programs in the "bin" directory. If these programs are not created, you should read the INSTALL document in the package directory. PREPARE INPUT FILE Our software requires only a binary input file (double-precision) of signal intensity data obtained during SNP genotyping. Before preparing the input file, please make sure that your intensity data is observed in the xy-coodinate and not transformed into the polar coordinate (there variables are often referred to as R and Theta). Our software does NOT allow signal intensities in polar coordinates. 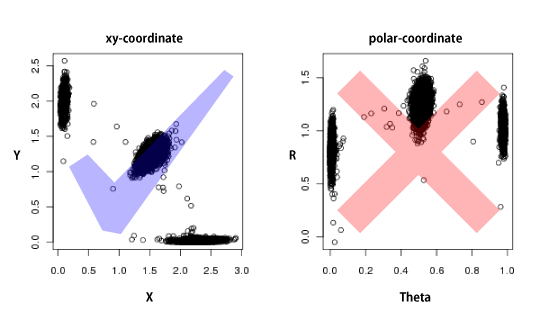 The file should be organized as an array of signal intensities in the xy-coordinate with length of 2 x L x M, where N is the sample size and L is the marker size (the number of loci). The order of intensities {x_ij, y_ij} for the subject i (i=1,...,N) at the locus j (j=1,...,L) in the input file is as follows: 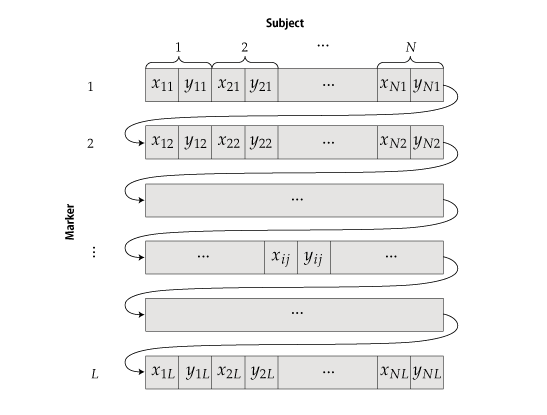 That is, the first 2N values are signal intensities of N subjects at the first locus, and the next 2N values are those at the second locus and so on. Note that, no line break nor space is needed so that the size of the binary file is 2 x N x L x sizeof(double) bytes. The use of our method is NOT limited to a specific genotyping platform (e.g., Illumina, Affymetrix, and so on). We only requare that the avarage of signal intensities for each subject is 2; Å@Å@Sum_{j=1}^L (x_ij + y_ij) / N = 2. This fact implies that the avaraged intensity reflects the chromosomal copy number of 2. Again, we note that (x_ij, y_ij) is observed in the xy-coordinate but not in the polar coordinate. Here the more sophisticated signal intensity corrections, such as the quantile normalization (Bolstad et al., 2003), GC correction by smoothing-spline and whole genome amplification correction by LOESS (Marioni, et al., 2007) are preferable for better fitting of the GMM. Note that the loci are not necessarily in physical order, because the model is fitted on a marker-by-marker basis; that is, we genotype the CNP for each marker independently across all the subjects (in the same manner as SNP genotyping). FIT BAYESIAN GMM BY gmm The command for fitting the Bayesian GMM is very simple. Here is an example command line: % gmm -i intensity.bin -o gmm.bin -n N > log The first argument is an input file name of the intensity file defined in the previous section. The second argument is an output file name and the third is the sample size. The user can rearrange the order of arguments as far as the option and value are coupled. For more detailed information on other options, see the help: % gmm -h CALL INDIVIDUAL'S GENOTYPES BY call The command for inferring allele-specific genotypes is also very simple. Here are some example command lines: % call -i intensity.bin -g gmm.bin -n N -o geno -a -c > log % call -i intensity.bin -g gmm.bin -n N -o geno -d > log % call -i intensity.bin -g gmm.bin -n N -o geno -m --hwep 1e-3 > log The first common argument is an input file name of the intensity file defined in the previous Section. The second common argument is the file name of the output file produced by gmm in the previous section and the third is the sample size. The forth common argument specifies the prefix of output files. The call program can provide the maximum a posteriori (MAP) estimation and the posterior mean estimation of copy number dosages and allelic variations (see our manuscript for the definition of estimators). The first line of the above examples indicates the call provides the MAP allele-specific copy number dosages (specified by -a option) along with MAP copy number dosages (specified by -c option). The second line provides the posterior mean CN dosages (specified by -m option). Note that you need to specify, at least, one of -a, -c -d, -l and -m options. The call program does not provide genotypes for all loci but only for the locus that meets strict QC thresholds. An output file with the suffix ".status" may let you know which locus was genotyped by call. The second column of the file indicates whether the locus was 1: called or 0: not called. Note that If you specify -f option, then call provides genotypes for all loci. You may also modify a part of the QC thresholds. The third line of the above examples indicates the P-value threshold of the Hardy-Weinberg equilibrium test is 1e-3 (1e-6 as default). For more detailed information on other options, see the help: % call -h VISUAL CONFIRMATION (OPTIONAL) We employ strict QC conditions to reduce false positive CNPs and genotyping errors. However, it is obvious that it is not 100% certain to avoid these situations. The only one thing you can do is to see the fitted GMM result by your eyes. Here we provide a useful functions in the R software to visualize the fitted GMM and infered genotypes along with signal intensity data. In the "R" directory, we present three scripts, "pe.R", "RGB.R" and "plot.R" which are used in the test run of our package (see INSTALL for details). The script "pe.R" provides a function of plotting each component of the GMM. The script "RGB.R" gives a color coding function for different copy number genotypes, which was used in our manuscript. The R script "plot.R" produces a series of plots of signal intensities for multiple loci along with the fitted GMMs and inferred copy number dosages. Note again, PlatinumCNV is a standalone package and independent of R. It does NOT require to install R. However we recommend you to see and confirm your result by eyes. We are sure that this extra procedure makes your result much more reliable. REFERENCES Kumasaka N., et al. (2011) PlatinumCNV: a Bayesian Gaussian mixture model for genotyping copy number polymorphisms using SNP array intensity data. Genet. Epidemiol. 35:8, 831-844. Bolstad, et al. (2003) A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 19, 185-93. Marioni, et al. (2007) Breaking the waves: improved detection of copy number variation from microarray-based comparative genomic hybridization. Genome Biology. 8, R228. ACKNOWLEDGEMENTS Thanks to Prof. Ryo Yamada at Kyoto University and Prof. Seiya Imoto at the University of Tokyo for their helpful suggestions and comments. We also thank Mr Kouhei Tomiduka and Mr Yoshiyuki Yukawa at NEC Informatec Systems, Ltd. for their kind support in developing the software package. This project was conducted as a part of the BioBank Japan Project. << HOME |