PCAj: Population Structure Prediction System for Japanese | ō·¢{īĻāyü[āW
 EXE (Windows) | JAR (Multi-platform) | Test Data
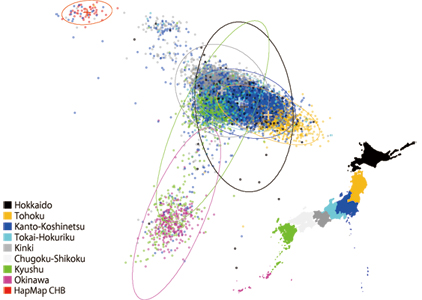
This application predicts population structure of Japanese
samples using genome-wide SNP genotypes. It creates a 2D
scatterplot of predicted principal components based on the
probabilistic PCA. It also provides posterior probabilities
from which an individual has descended from each of several
Japanese ancestries:
- Hokkaido
- Tohoku
- Kanto-Koshinetsu
- Tokai-Hokuriku
- Kinki
- Kyushu
- Okinawa
based on the result of LDA. The application is based on
SNP markers included in the Illumina HumanHap 550K chip.
However you may use SNP genotype data observed from other
platforms as far as a set of SNPs overlaps with that of the
550K chip. Note that the more SNPs you input, the better
prediction result you get (at least 10,000 SNPs
are recommended).
usage: <genotype_file>
Example: java -jar pca.jar test.txt
The <genotype_file> parameter should be a file that contains
SNP genotype data. The first column of the data is SNP rs_ID
which should be sorted as dictionary order. The second column
is SNP genotypes for all subjects in the sample set without
any separator. The first and second columns should be TAB
separated. Here genotypes should be encoded as 0, 1 and 2
for a homozygote pair of A/T, a heterozygote pair of A/T and C/G
and the other homozygote pair of C/G, respectively. The missing
genotype should be encoded as 9.
Example: AC, AA, CC, TT, AG, GG, TC, NN -> 1, 0, 2, 0, 1, 2, 1, 9
For Windows users, you can drag & drop your data onto the icon of
the execulable file (pca.exe).
If you use this software for publication, please send a note
with the reference or a link. When citing this software, use:
Kumasaka et al. (2010) Establishment of a Standardized System
to Perform Population Structure Analyses with Limited Sample
Size or with Different Sets of SNP Genotypes.
Journal of Human Genetics, 55(8):525-33.
(c) Natsuhiko Kumasaka (kumasaka AT src.riken.jp)
http://kumasakanatsuhiko.jp/projects/popstr/
EXE (Windows) | JAR (Multi-platform) | Test Data
This application predicts population structure of Japanese
samples using genome-wide SNP genotypes. It creates a 2D
scatterplot of predicted principal components based on the
probabilistic PCA. It also provides posterior probabilities
from which an individual has descended from each of several
Japanese ancestries:
- Hokkaido
- Tohoku
- Kanto-Koshinetsu
- Tokai-Hokuriku
- Kinki
- Kyushu
- Okinawa
based on the result of LDA. The application is based on
SNP markers included in the Illumina HumanHap 550K chip.
However you may use SNP genotype data observed from other
platforms as far as a set of SNPs overlaps with that of the
550K chip. Note that the more SNPs you input, the better
prediction result you get (at least 10,000 SNPs
are recommended).
usage: <genotype_file>
Example: java -jar pca.jar test.txt
The <genotype_file> parameter should be a file that contains
SNP genotype data. The first column of the data is SNP rs_ID
which should be sorted as dictionary order. The second column
is SNP genotypes for all subjects in the sample set without
any separator. The first and second columns should be TAB
separated. Here genotypes should be encoded as 0, 1 and 2
for a homozygote pair of A/T, a heterozygote pair of A/T and C/G
and the other homozygote pair of C/G, respectively. The missing
genotype should be encoded as 9.
Example: AC, AA, CC, TT, AG, GG, TC, NN -> 1, 0, 2, 0, 1, 2, 1, 9
For Windows users, you can drag & drop your data onto the icon of
the execulable file (pca.exe).
If you use this software for publication, please send a note
with the reference or a link. When citing this software, use:
Kumasaka et al. (2010) Establishment of a Standardized System
to Perform Population Structure Analyses with Limited Sample
Size or with Different Sets of SNP Genotypes.
Journal of Human Genetics, 55(8):525-33.
(c) Natsuhiko Kumasaka (kumasaka AT src.riken.jp)
http://kumasakanatsuhiko.jp/projects/popstr/
|